MULTI
Integration of multi-omics data of microbial species
Short description
Many microbiome researchers seek to investigate the functional potential of microbial ecosystems with a holistic, multi-omics approach. Currently, interoperability between genomic, transcriptomic, and proteomic data for instance is a bottleneck. The workflow of this analysis that is being established by MULTI tackles such issues to facilitate future work for members of the community.
The first application and basis for the establishment of the workflow is the investigation of antimicrobial resistance in different Listeria monocytogenes strains by applying multi-omics data analysis. Three different L. monocytogenes strains were cultured on sub-lethal concentrations of biocides, their genomes sequenced, and transcriptomics and proteomics data collected. These data will provide insights into the functional responses of the bacteria to the biocide by analyzing pathways that differ in their gene expression or protein translation. Genome-scale metabolic reconstructions can be integrated with both transcriptomics and proteomics data, resulting in multi-omics data integration, which can provide insights into the different functional responses of microbial cells to an antimicrobial substance.
Graphical abstract
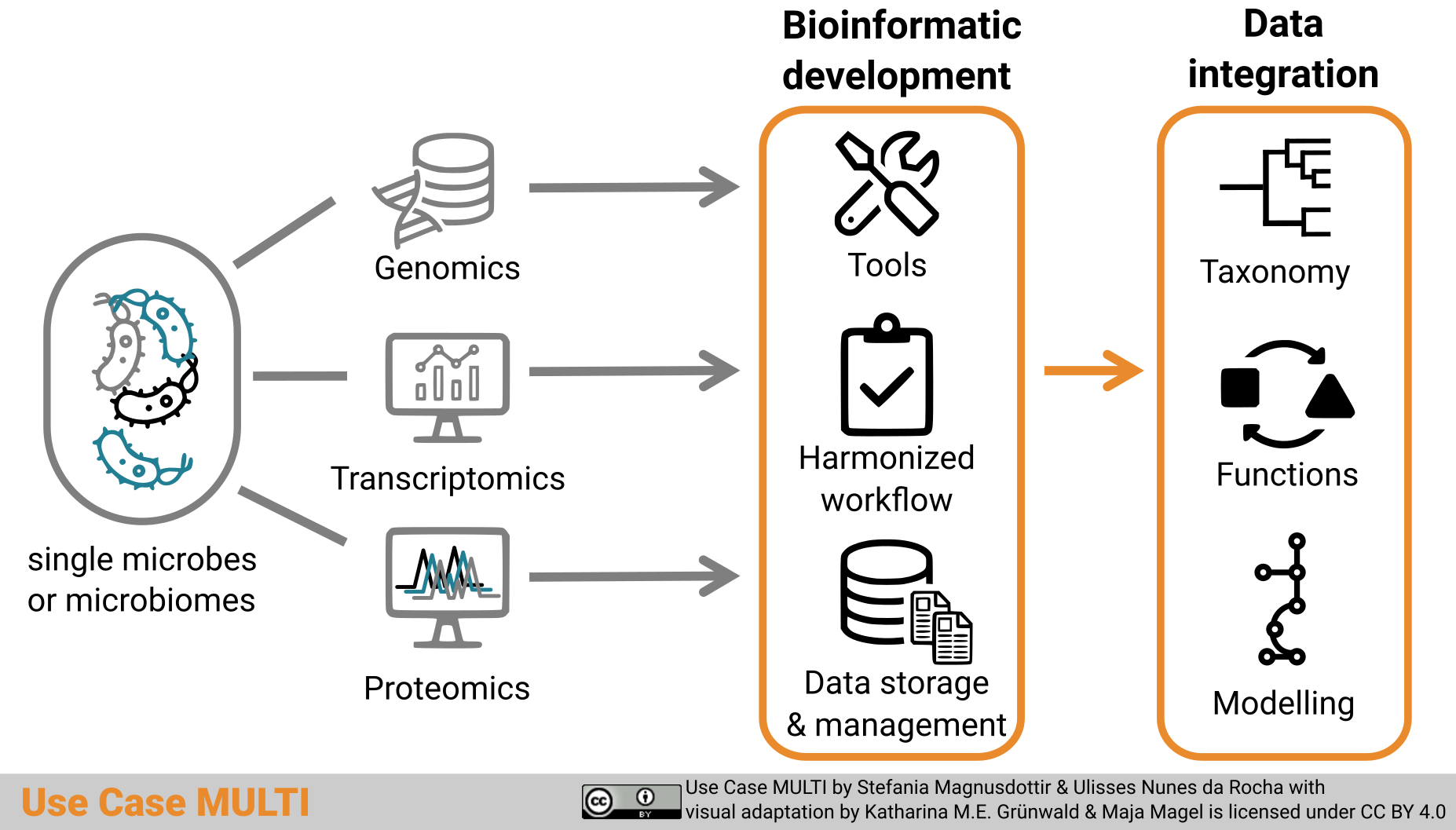
Graphical abstract Use Case MULTI by Stefania Magnusdottir and Ulisses Nunes da Rocha with visual adaptation by Katharina M.E. Grünwald and Maja Magel is licensed under a Creative Commons Attribution 4.0 International License CC BY 4.0.
How you can contribute
You are a …
- microbiologist with short and long read genomic sequences of bacterial isolates:
Use the workflow to assemble circular genomes of your isolates, annotate genes and proteins, and create genome-scale metabolic reconstructions. - microbiologist with isolate genome sequences and transcriptomics and/or proteomics data:
Use the workflow to analyze differential gene expression (transcriptomics), protein translation (proteomics), and integrate these data sets with the genomes using genome-scale metabolic reconstructions. - microbiologist with experience in transcriptomics or proteomics analyses:
Check out the preliminary guidelines on data processing, analysis, and integration of genomics, transcriptomics, and proteomics data.
Give suggestions and feedback on the workflow and useful additional features that could be implemented.
Planned output
Modular analysis pipeline
Creating a modular pipeline on multi-omics analyses on closely-related bacterial strains or species
Selected state-of-the-art tools and scripts for genome circularization and annotation, differential expression RNA seq analysis (transcriptomics), automatic genome-scale metabolic reconstruction, and context-specific metabolic models from transcriptomics data
Create a modular pipeline on multi-omics analyses on non-closely-related bacterial strains or species based on the previous pipeline
Create a modular pipeline on multi-omics analyses of microbial communities
Selected state-of-the-art tools and scripts for MAG assembly from metagenomic data, taxonomic lineage assignment, MAG quality control
Selected a state-of-the-art tool for hybrid genome assembly using short and long genomic sequences.
Select a state-of-the-art tool for proteomics analysis.
Infrastructure output
- Create a workflow that can facilitate the integration and analysis of multi-omics data by ensuring that the inputs and outputs of each tool are compatible with each other.
- Create written and video tutorials to train researchers to use the workflow and the individual tools therein
Achievements
- Created a guideline for data processing, analysis, and integration of genomics, transcriptomics, and proteomics data:
https://gitfront.io/r/user-6306825/uUtkLPVoZfbX/multi/ - MultiPath is a nextflow-port of the NFDI4Microbiota MULTI Use Case workflow for the integration of multi-omics data of microbial species: https://github.com/grp-bork/MultiPath and https://clowm-staging.bi.denbi.de/
BioDeepFuse
A total of 1879 metagenome-assembled genomes (MAGs) were recovered using MuDoGeR from the metagenomic data, forming the basis for transcriptome mapping to determine whether non-coding RNAs (ncRNAs) were not transcribed and to provide insights into the genetic and functional composition of the microbial ecosystems. A non-coding RNA database was established using data from 23,000 MAGs from the CLUETERRA, creating a comprehensive resource for advanced transcriptomic and multi-omics research.
BioDeepfuse is a hybrid deep learning framework that integrates CNN/BiLSTM models with handcrafted feature extraction techniques, enabling superior non-coding RNA classification by effectively capturing spatial and sequential patterns in RNA sequences.
- Publication on BioDeepFuse: a hybrid deep learning approach with integrated feature extraction techniques for enhanced non-coding RNA classification: https://doi.org/10.1080/15476286.2024.2329451
- BioDeepFuse tool: https://github.com/brenoslivio/BioDeepFuse
<< Back to All Use Cases
Project Lead

Anderson Paulo Avila Santos
ORCID ID: 0000-0002-6791-0768
Helmholtz Centre for Environmental Research (UFZ), Department of Environmental Microbiology (UMB)

Stefania Magnusdottir (former)
ORCID ID: 0000-0001-6506-8696
Helmholtz Centre for Environmental Research (UFZ), Department of Environmental Microbiology (UMB)
Ulisses Nunes da Rocha
ORCID ID: 0000-0001-6972-6692
Helmholtz Centre for Environmental Research (UFZ), Department of Environmental Microbiology (UMB)
Keywords
multi-omics
genomics
transcriptomics
proteomics
genome-scale metabolic reconstruction