MockBench
Systematic assessment in metagenomics using high diversity Mock samples
Short description
Microbiologists and bioinformaticians often struggle to identify the strengths and weaknesses of different protocols, sequencing techniques or analysis approaches in the field of metagenomics. This issue can be addressed by benchmarking studies that rely on well-characterized benchmarking datasets and thus can give advice on standards and best practices. Often, these datasets are established through the generation of artificial microbial Mock communities. While existing Mock datasets moved the field forward, many accumulated flaws: missing laboratory protocols, insufficient controls, outdated sequencing technology, and limited species diversity to suit real-life evaluation scenarios.
Therefore, the main goal of MockBench is to create new, highly diverse Mock samples based on strains covering the entire tree of bacterial life, sequenced with Next-generation sequencing (NGS) instruments (MiSeq amplicon sequencing, Illumina Whole-Genome Sequencing (WGS), Oxford Nanopore long-read amplicons), while being quality controlled and well documented. This will create a FAIR and metadata-rich gold standard that can be used in all metagenomic evaluations and benchmarking efforts and eventually be integrated into NFDI4Microbiota and ELIXIR benchmarking services, e.g. the OpenEBench platform.
Graphical abstract
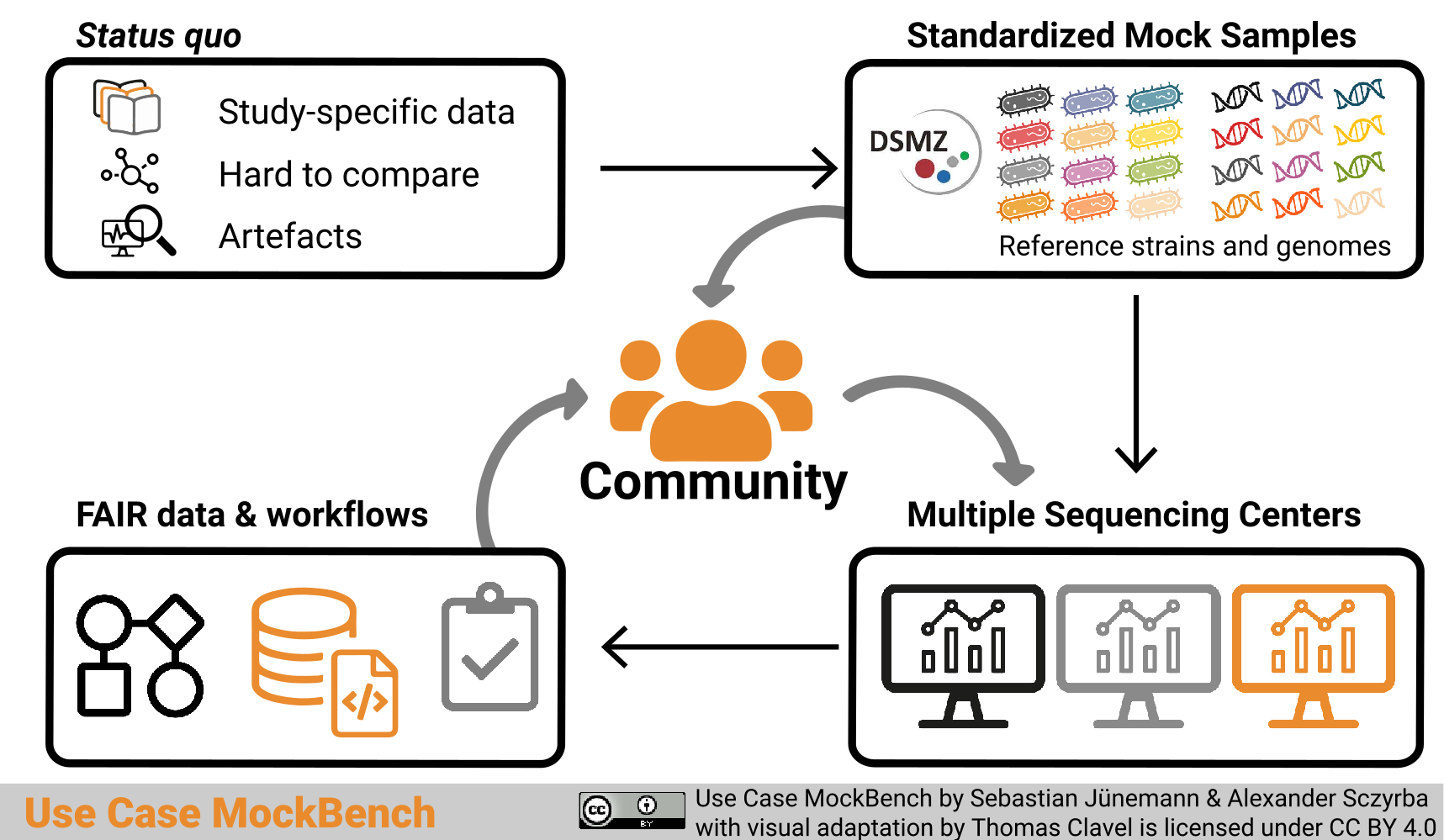
Graphical abstract “Use Case MockBench” by Sebastian Jünemann and Alexander Sczyrba with visual adaptation by Thomas Clavel is licensed under a Creative Commons Attribution 4.0 International License (CC BY 4.0).
How you can contribute
You are a …
- bioinformatic software developer interested in evaluating your tools:
Specify the application scenario for which your tool is most likely to be used and, consequently, indicate what challenges are to be expected from a typical target microbial community in terms of complexity and diversity. What key parameters should an appropriate evaluation dataset feature? - bioinformatician interested in benchmarking different tools:
Describe what sources for data acquisition are known or already used for the benchmarking and to what extent requirements for metadata descriptions and FAIR data deposition is a factor to that. Especially for larger benchmarks, outline how data transfer is managed and what technologies are being used to conduct the benchmark, e.g. object storage and cloud technologies on Nextflow workflows. - microbiologist expert with metagenomic data:
Describe your preferred analysis platform or collection of software tools along with the reason for your selection. Microbiologists who are interested in appropriate tool selection based on focused benchmarks or general advice on appropriate tool selection will be trained on how to perform benchmarks on their own. - microbiologist beginner with metagenomic data:
Get trained on and use the automated analysis pipeline. Let us know the major issues you face when dealing with and analyzing metagenomic data. - microbiologist experienced in or with access to sequencing technologies:
Contribute by applying alternative protocols for the different library preparation steps on the genomic Mock community and/ or by conducting sequencing runs of the prepared sequencing libraries, allowing us (and you) to compare sequencing efforts and procedures.
Planned output
Research
- Developed and established methods on the creation of a Mock sample
- Preparing and publishing FAIR and metadata rich Mock datasets
- Deposit FAIR and easily integrable Mock dataset to be used in custom evaluations and benchmarks
- Provide benchmark results of different amplicon analysis methods
- Compare targeted and whole genome shotgun metagenomics based on the generated Mock sample
Infrastructure output
- Create training material on amplicon analyses, workflows, and the application of Mock samples for benchmarking for an annual training course on metagenomic analysis
- Establish an inventory of current processes on metadata standards, metadata collection, and their integration with analysis tools
- Developing the analysis workflows with Nextflow in compliance with NFDI4Microbiota standards and guidelines
- Implementing the workflows in the de.NBI Cloud environment
Project Lead

Sebastian Jünemann
ORCID ID: 0000-0002-3714-0328
Institute for Bio- and Geosciences, Computational Metagenomics (IBG-5), Research Center Jülich, Jülich, c/o Centrum für Biotechnologie (CeBiTec)

Alexander Sczyrba
ORCID ID: 0000-0002-4405-3847
Bielefeld Institute for Bioinformatic Infrastructure, Computational Metagenomics, Faculty of Technology, Bielefeld University, Bielefeld
Keywords
metagenomics
targeted metagenomics
amplicons
mock community
mock dataset
benchmarks
workflows